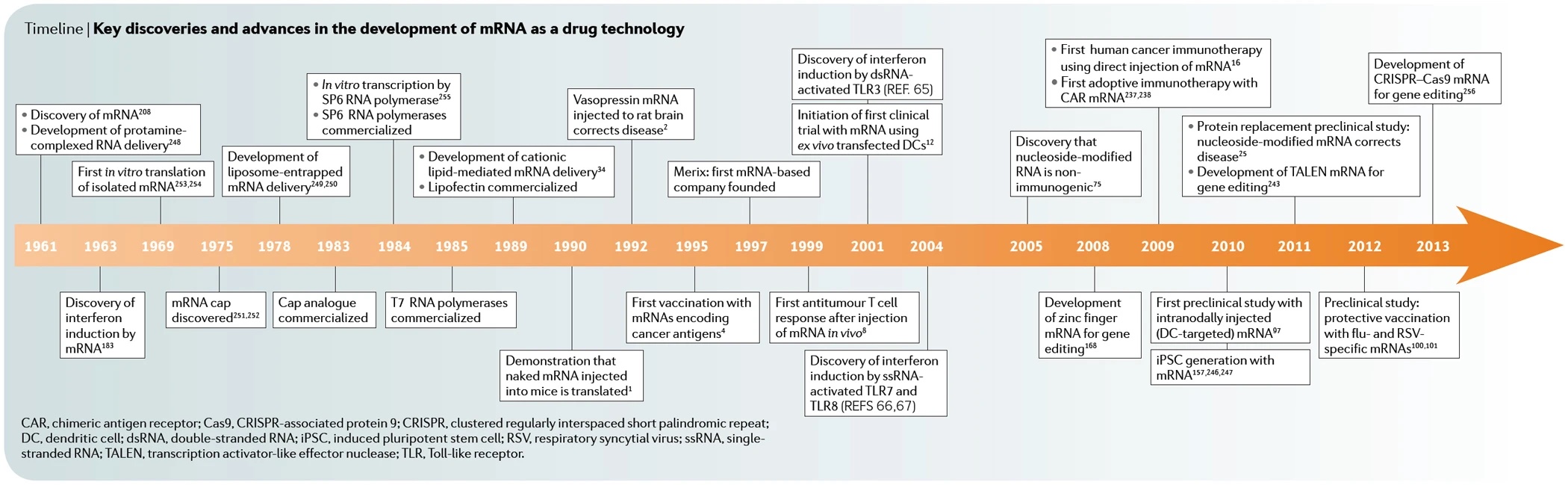
")
")

4 November 2021
Nach der Zulassung der weltweit ersten Gentherapie für eine bestimmte Form der Retinitis pigmentosa (RP) in 2017 berichten Forscher nun über einen weiteren gentherapeutischen Ansatz zur Behandlung dieser Krankheit unter Verwendung von Optogenetik. Mediziner aus den USA und der Schweiz konnten einem 58-jährigen Mann, der durch die Netzhaut-Erkrankung seit Jahrzehnten blind war, dazu verhelfen, wieder Gegenstände erkennen und greifen zu können.
So erkannte der Patient mithilfe einer Spezialbrille ein Telefon, eine Tür oder einen Zebrastreifen, wie das Team um José-Alain Sahel von der University of Pittsburgh School of Medicine und Botond Roska von der Universität Basel in Nature Medicine im Mai 2021 beschrieb.
Bei der RP sind bestimmte Gene mutiert, die für Licht-Rezeptoren in der Netzhaut kodieren. Es sind mehr als 70 Gene bekannt, deren Nicht-Funktion zur Folge hat, dass die Sehfähigkeit allmählich zugrunde geht. Laut den Autoren sind weltweit über zwei Millionen Menschen betroffen, in Deutschland sind es geschätzt bis zu 40.000. Gegen die vererbte Krankheit gibt es bislang nur eine zugelassene Therapie. Diese bezieht sich lediglich auf frühe RP, die durch eine Mutation im Gen RPE65 ausgelöst wird und bei der die Lichtrezeptor-Zellen noch nicht abgestorben sind.
Weltweit erste RP-Gentherapie
Im Dezember 2017 ließ die US-Behörde FDA die erste Gentherapie für eine erblich bedingte Augen-Erkrankung, die bereits im Alter von 20 Jahren zur Erblindung führen kann, zu. Entwickelt wurde LUXTURNA (Voretigene neparvovec) von der 2013 gegründeten US-Biotech-Firma Spark Therapeutics. 2018 folgte die Zulassung in Europa durch die EMA (European Medicines Agency). Novartis aus der Schweiz sicherte sich globale Vermarktungs-Rechte - mit Ausnahme der USA.

Wirkmechanismus von Luxturna
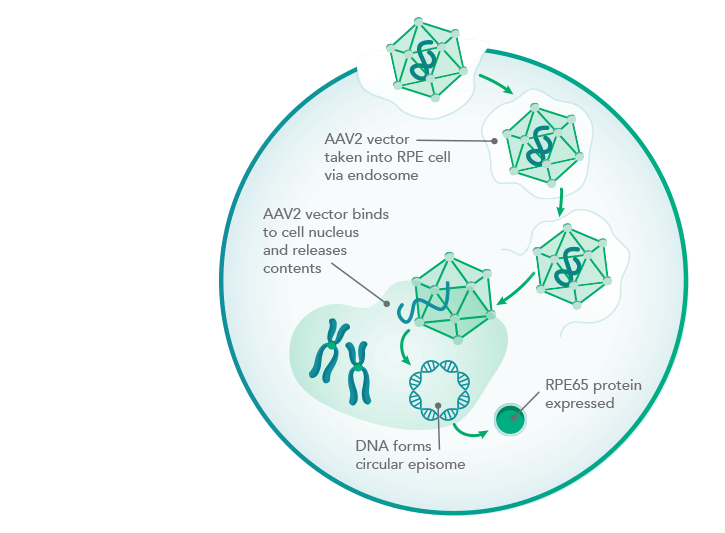
Wirkmechanismus von Luxturna
Im Dezember 2019 bezahlte die ebenfalls in der Schweiz beheimatete Roche 4,3 Milliarden US$ für die Übernahme von Spark, die als unabhängige Tochterfirma bestehen bleibt.
LUXTURNA nutzt als Genfähre einen Virus, der die Retina-Zellen mit einer funktionsfähigen Kopie vom RPE65-Gen versorgt. Das von diesem kodierte RPE65-Protein ist ein bei der Krankheit fehlendes Seh-Pigment. Mit einem Preis von 850.000 US$ zählt die Therapie zu der teuersten auf dem US-Markt.
Was ist Optogenetik und wie therapiert sie Blinde?
Bei der Optogenetik geht es grundsätzlich darum, genetische Informationen in Ziel-Zellen einzuschleusen, die für lichtempfindliche Ionen-Kanäle, Transporter oder Enzyme kodieren. Letztendlich sind dadurch zelluläre Aktivitäten mit Licht zu steuern.
Ausgangspunkt waren Forschungen vom Team um Peter Hegemann von der Humboldt-Universität in Berlin, das sich in den 1990er mit der Grünalge Chlamydomonas beschäftigte und auf sogenannte Kanal-Rhodopsine stieß. Diese Proteine wirken in bestimmten Zellen wie Schalter, die auf Licht reagieren.
2004 schaffte es das Team um Karl Deisseroth von der Stanford University das Gen dieser Algenproteine in Nerven-Zellen einer Maus einzubauen. Sie produzierten daraufhin das Protein und wurden somit lichtempfindlich. Es war damit möglich, durch Licht-Impulse die Maus fernzusteuern: Sie lief im Kreis herum, stoppte und rannte wieder los.
Seither gilt Deisseroth als der Begründer der Optogenetik, also der Steuerung genetisch veränderter Zellen mithilfe von Licht. Die Technik wurde von Nature Methods zur Methode des Jahres 2010 gekürt.
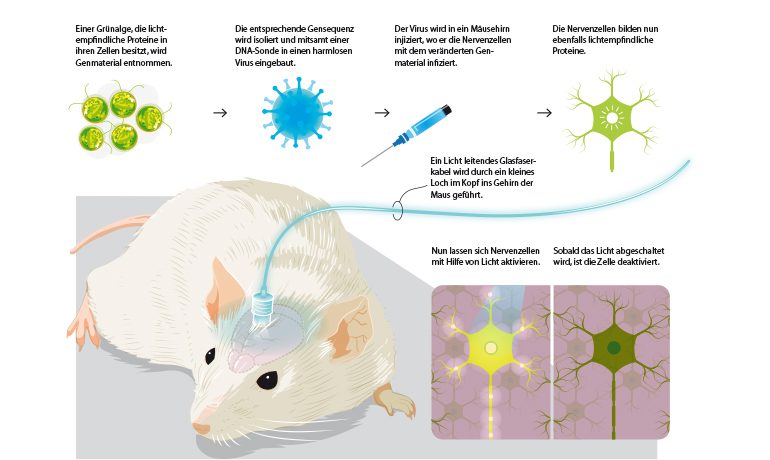
Mittels eines in das Hirn eingeführten Glasfaserkabels können im Maus-Versuch Nervenzellen aktiviert oder deaktiviert werden. Bildquelle: KlarText
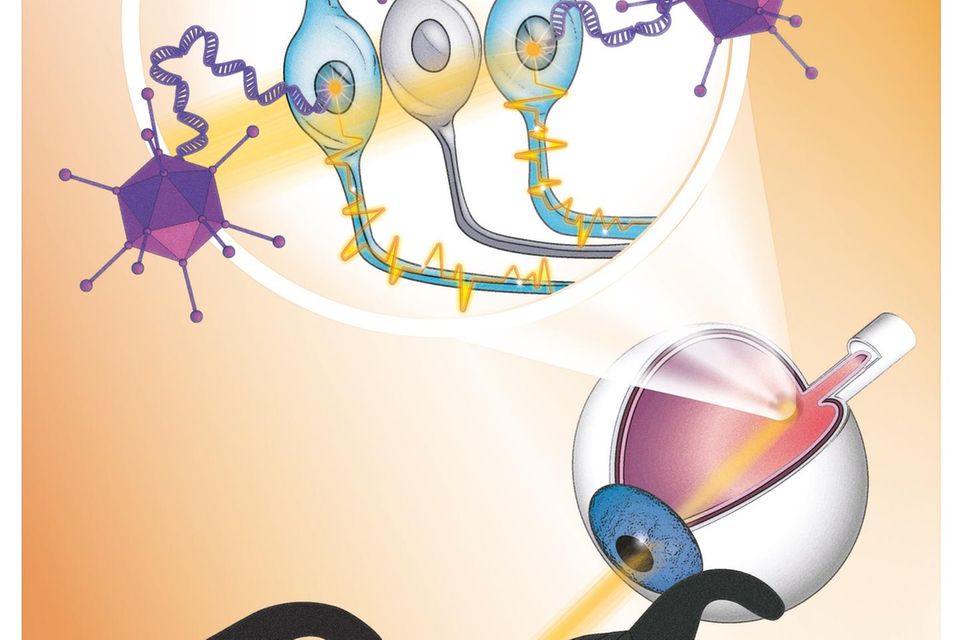
Die neue Gentherapie macht die Netzhaut eines Blinden lichtempfindlich, unterstützt durch eine lichtstimulierende Schutzbrille, die mit einer neuromorphen Kamera Bilder aus der visuellen Welt aufnimmt. Bildquelle: GEO
Für die Therapie des blinden Patienten bietet die Optogenetik den Vorteil, dass sie mutations-unabhängig einsetzbar ist. Denn es werden neue lichtempfindliche Moleküle ins Auge gebracht und nicht versucht, nicht-funktionierende durch einen Gen-Transfer zu ersetzen.
In der Studie injizierte das Team in das schwerer betroffene Auge des Mannes einmalig Viren, die den Bauplan für das lichtempfindliche aus Mikroalgen stammende Protein (Kanalrhodopsin) übertrugen. Dieses bildet in bestimmten Zellen der unteren Netzhaut einen Ionenkanal. Der wird benötigt, um aus einfallendem Licht elektrische Signale zu erzeugen, die die Zellen an das Sehzentrum im Gehirn weiterleiten.
Da Tageslicht nicht ausreicht, um den Ionenkanal zu aktivieren, unterstützt eine Kamerabrille, indem sie Bilder der Umgebung in Echtzeit mit sehr hoher Intensität durch die Pupille auf die Netzhaut projiziert.
Der Patient begann ein Training mit der Brille viereinhalb Monate nach der Injektion. Das war der Zeitraum, den die Zielzellen benötigten, um in der unteren Netzhaut Ionenkanäle zu bilden. Nach sieben Monaten berichtete er dann von visuellen Eindrücken, allerdings nur beim Tragen der Brille. Die behandelnden Mediziner warnen jedoch vor überzogenen Hoffnungen. Derzeit sei nicht zu erwarten, dass Patienten nach einer solchen Therapie etwa Gesichter erkennen oder gar lesen könnten. Dennoch ermögliche das Verfahren eine deutliche Steigerung der Lebensqualität - zumal der 58-Jährige nur mit einer niedrigen Dosis behandelt worden sei.
Peter Hegemann spricht von einem "Durchbruch in eine neue Ära zur Behandlung von Retinitis pigmentosa". Der klinische Einsatz sei jedoch "noch lange nicht absehbar und vieles kann in naher Zukunft verbessert werden", betont der Berliner Neuro-Wissenschaftler, der zu den Entdeckern der Kanalrhodopsine zählt.
Verwendete Quellen:
- Artikel in Geo vom 25. Mai 2021
- Artikel in der Süddeutsche Zeitung vom 25. Mai 2021
- Original-Artikel von Sahel et al., Partial recovery of visual function in a blind patient after optogenetic therapy, Nature Medicine, online veröffentlicht am 24. Mai 2021
- Artikel in KlarText, Zellen steuern mit Licht – und Liebe, Einschub von Joachim Schüring